




Il Regolamento (UE) 2017/745 sui dispositivi medici (MDR) stabilisce requisiti specifici riguardanti le informazioni che i fabbricanti devono fornire insieme ai loro dispositivi, con particolare attenzione alle esigenze linguistiche. Questi requisiti sono fondamentali per garantire che le informazioni siano comprese dagli utilizzatori finali, siano essi professionisti sanitari o pazienti (utilizzatori profani).
Obblighi dei fabbricanti secondo l’Articolo 10 del MDR
L’Articolo 10 del MDR delinea gli obblighi generali dei fabbricanti, tra cui:
- progettazione e fabbricazione conformi à i fabbricanti devono garantire che i dispositivi siano progettati e fabbricati in conformità alle prescrizioni del regolamento;
- sistema di gestione del rischio à è obbligatorio istituire, documentare, e mantenere un sistema per la gestione del rischio come descritto nell’allegato I del MDR, punto 3;
- documentazione tecnica à è necessario redigere e mantenere aggiornata la documentazione tecnica in modo da consentire che sia valutata la conformità del dispositivo alle prescrizioni del regolamento (allegato II).
Inoltre, i fabbricanti provvedono a che il dispositivo sia corredato delle informazioni indicate all’allegato I del MDR, punto 23 “Etichette e istruzioni per l’uso”, in una delle lingue ufficiali dell’Unione stabilita dallo Stato membro in cui il dispositivo è messo a disposizione dell’utilizzatore o del paziente. Le indicazioni che figurano sull’etichetta devono essere indelebili e scritte in modo da risultare facilmente leggibili e chiaramente comprensibili all’utilizzatore o al paziente previsto.
Requisiti linguistici secondo l’allegato I MDR, punto 23 “Etichette e istruzioni per l’uso”
Il Capo III dell’allegato I del MDR “Requisiti riguardanti le informazioni fornite con il dispositivo” specifica i requisiti da soddisfare relativamente alle informazioni fornite con il dispositivo, tra cui etichettate e istruzioni per l’uso (punto 23). È essenziale che queste informazioni siano fornite nella lingua o nelle lingue ufficiali degli Stati membri in cui il dispositivo è commercializzato, come stabilito dalle Autorità Competenti nazionali.
La Commissione Europea, in collaborazione con gli Stati membri, ha pubblicato una linea guida che, sottoforma di tabella, identifica i requisiti linguistici specifici per ciascun paese. Questo documento mira ad assistere i fabbricanti nell’identificazione delle lingue accettate dai diversi Stati membri per la redazione delle informazioni (etichette e istruzioni per l’uso) che accompagnano un dispositivo immesso in commercio nei paesi dell’Unione Europea.
Il documento “MDR – Language requirements for manufacturers” è disponibile al seguente link: Overview of language requirements for manufacturers of medical devices.
Allo stesso link è possibile trovare le informazioni pertinenti per il Regolamento (UE) 2017/746 (IVDR) relativamente ai Dispositivi Medico-Diagnostici in vitro.
Requisiti per l’Italia
La normativa di riferimento in Italia è il Decreto Legislativo 5 agosto 2022, n.137 “Disposizioni per l’adeguamento della normativa nazionale alle disposizioni del regolamento (UE) 2017/745 del Parlamento europeo e del Consiglio, del 5 aprile 2017, relativo ai dispositivi medici, che modifica la direttiva 2001/83/CE, il regolamento (CE) n. 178/2002 e il regolamento (CE) n. 1223/2009 e che abroga le direttive 90/385/CEE e 93/42/CEE del Consiglio, nonché’ per l’adeguamento alle disposizioni del regolamento (UE) 2020/561 del Parlamento europeo e del Consiglio, del 23 aprile 2020, che modifica il regolamento (UE) 2017/745 relativo ai dispositivi medici, per quanto riguarda le date di applicazione di alcune delle sue disposizioni ai sensi dell’articolo 15 della legge 22 aprile 2021, n. 53” (Decreto Legislativo 5 agosto 2022, n. 138 per quanto riguarda i dispositivi medico-diagnostici in vitro).
Il decreto specifica, all’articolo 6, che “Le informazioni e le indicazioni relative a qualsiasi tipologia di dispositivo medico, fornite per iscritto dal fabbricante all’utilizzatore e al paziente … sono espresse in lingua italiana al momento della consegna all’utilizzatore finale, per uso professionale o per qualsiasi altra utilizzazione.”
La linea guida “MDR – Language requirements for manufacturers” – menzionata precedentemente – fornisce indicazioni circa gli obblighi specifici per l’Italia, riportando i seguenti requisiti:
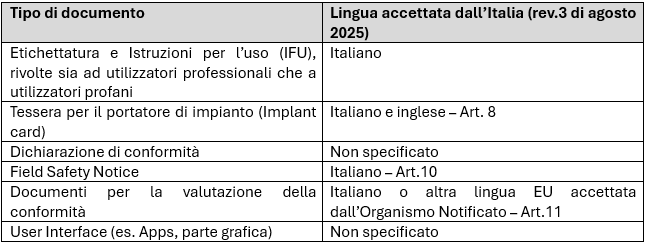 Ulteriori aggiornamenti potrebbero essere introdotti dalla Commissione Europea, pertanto è consigliabile monitorare costantemente l’evoluzione dei requisiti o consultare un esperto per effettuare le opportune verifiche.
Ulteriori aggiornamenti potrebbero essere introdotti dalla Commissione Europea, pertanto è consigliabile monitorare costantemente l’evoluzione dei requisiti o consultare un esperto per effettuare le opportune verifiche.
Considerazioni per i fabbricanti
La seguente lista include le attività utili per garantire la conformità dei dispositivi medici ai requisiti del MDR:
- verificare i requisiti linguistici nazionali à consultare le linee guida aggiornate fornite dalla Commissione Europea per comprendere le specifiche esigenze linguistiche di ciascun paese membro.
- adeguare la documentazione à assicurarsi che etichettatura, istruzioni per l’uso e altre informazioni correlate siano tradotte accuratamente nelle lingue richieste, tenendo conto delle specificità terminologiche e culturali.
- mantenere aggiornamenti continui à monitorare eventuali modifiche normative o aggiornamenti dei requisiti linguistici per garantire una conformità costante.
Conclusioni
In conclusione, la conformità ai requisiti linguistici stabiliti dal MDR e dalle autorità nazionali è fondamentale per garantire la sicurezza e l’efficacia dei dispositivi medici nel mercato europeo. I fabbricanti devono prestare particolare attenzione a questi requisiti, adeguando le informazioni fornite con i dispositivi alle esigenze linguistiche dei diversi Stati membri.























