




Nell’ambito del Regolamento relativo ai Dispositivi Medici (MDR) 2017/745 uno studio si qualifica come “indagine clinica” se è di natura sistematica, coinvolge uno o più soggetti umani e è intrapreso per valutare la sicurezza o le prestazioni di un dispositivo (Articolo 2(45)). Le indagini cliniche non sono una novità introdotta dal MDR; tuttavia, quest’ultimo, rispetto alla Direttiva, sottolinea la necessità che il fabbricante di dispositivi medici raccolga dati clinici utili a garantirne la conformità, rafforzando gli obblighi documentali, clinici ed etici a cui devono adempiere le indagini cliniche, e fornendo maggiori dettagli su “cosa” è richiesto e su “come” fare.
Per la prima volta, l’MDR stabilisce il ruolo formale dello sponsor, definito come qualsiasi persona, società, istituzione oppure organizzazione che si assume la responsabilità di avviare, gestire e curare il finanziamento dell’indagine clinica (articolo 2 (49)). Nel caso in cui quest’ultimo non abbia una sede nell’Unione Europea, dovrà individuare un legale rappresentante sul territorio che sarà il responsabile e il garante del rispetto degli obblighi previsti per lo sponsor stesso.
Gli articoli dal 62 al 82 del nuovo MDR affrontano il “cosa”, ovvero le condizioni che devono essere soddisfatte per l’esecuzione di un’indagine clinica: coinvolgimento di un Comitato Etico e/o di un’Autorità Competente, la necessità di un consenso informato, le considerazioni sulle popolazioni vulnerabili, la procedura di richiesta, i requisiti per la conduzione dell’indagine clinica, registrazione e segnalazione degli eventi avversi, ecc. L’allegato XV specifica il “come”, stabilendo i requisiti minimi e la documentazione da presentare per la conduzione dell’indagine clinica, in particolare:
- presentazione della domanda di indagine clinica
- dossier dello sperimentatore
- piano di indagine clinica
- informazioni aggiuntive
- obblighi dello sponsor
- relazione sull’indagine clinica.
Da un punto di vista regolatorio, possono configurarsi due tipologie di indagini cliniche:
- Pre-market: necessarie al fine di una immissione in commercio in conformità ai requisiti regolamentari. Tali indagini vengono condotte con dispositivi privi di marcatura CE, o marcati CE ma modificati in modo sostanziale oppure marcati CE ma impiegati per una destinazione d’uso diversa da quella certificata.
- Post-market: condotte con dispositivi marcati CE, utilizzati secondo l’indicazione d’uso prevista, per confermare la sicurezza e le prestazioni del dispositivo, l’accettabilità del rapporto rischio/beneficio, l’identificazione del rischio residuo o dell’eventuale rischio emergente sulla base dell’impiego su larga scala o a lungo termine.
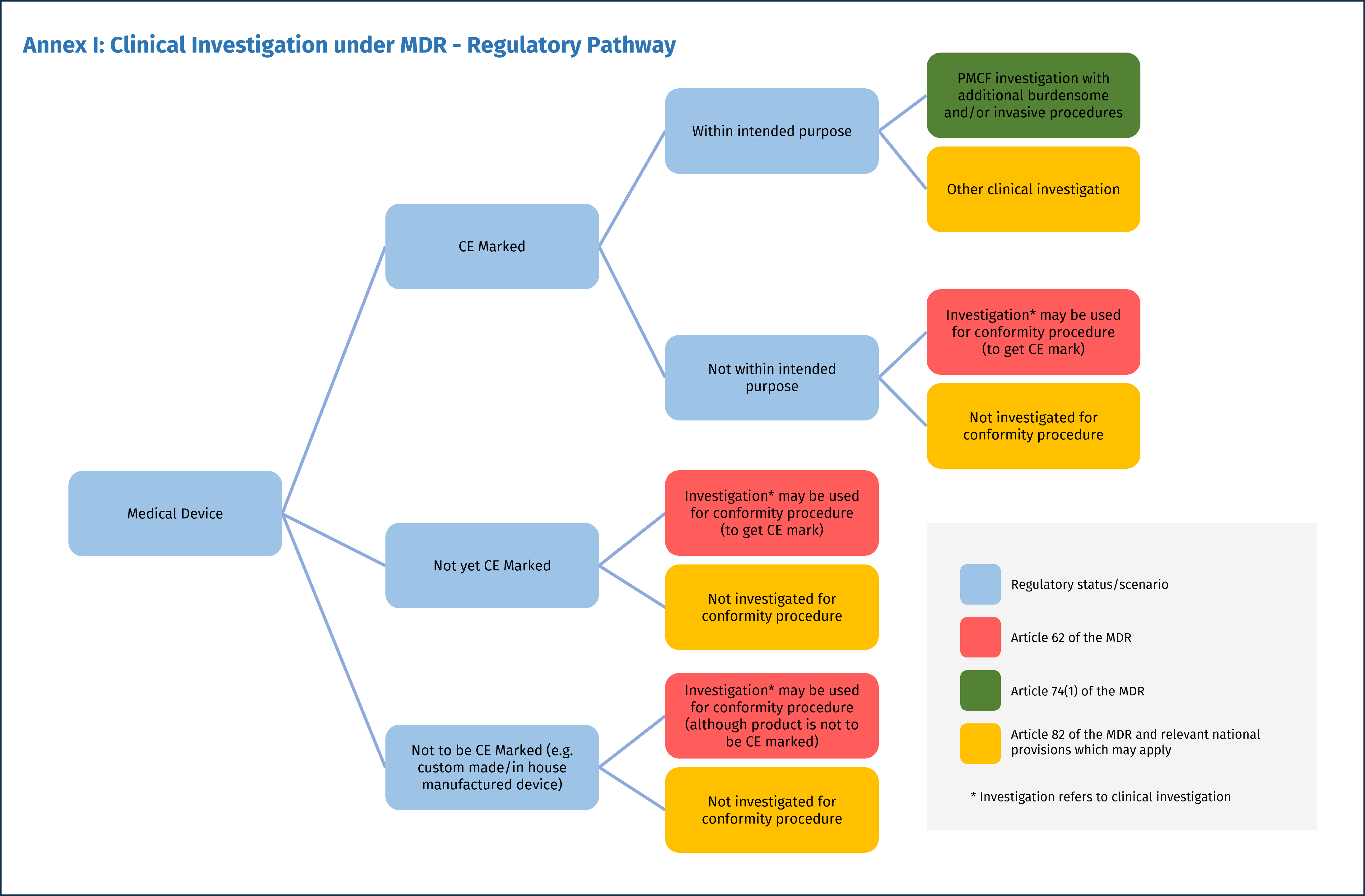
A seconda dello scenario regolatorio, l’indagine clinica del dispositivo può afferire a tre quadri normativi diversi, a cui corrispondono specifici requisiti e procedure (Figura 1):
- Le indagini cliniche condotte per stabilire la conformità del dispositivo dovranno seguire l’articolo 62
- Le indagini cliniche per la valutazione ulteriore della conformità(Post-Market Clinical Follow-up, PMCF), con procedure supplementari invasive o gravose rispetto a quelle eseguite nelle normali condizioni d’uso del dispositivo, saranno disciplinate dall’articolo 74(1)
- Per tutte le altre indagini cliniche con dispositivi medici condotte non ai fini della dimostrazione di conformità, troverà applicazione l’articolo 82.
(Clicca sulla tabella sottostante per ingrandirla.)
In ultima analisi, le indagini cliniche rappresentano una delle sfide più impegnative e difficili da affrontare, ma sono essenziali per dimostrare la sicurezza, il beneficio clinico o il valore aggiunto di un dispositivo medico. Oltre al MDR, diverse norme e linee guida europee possono essere prese in esame da fabbricanti, sponsor, Comitati Etici, ed ulteriori figure coinvolte nella progettazione e conduzione di un’indagine clinica. Tra queste, segnaliamo:
- ISO 14155:2020 Clinical investigation of medical devices for human subjects – Good clinical practice;
- MDCG 2021-6 Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation;
- MDCG 2021-08 Clinical investigation application/notification documents;
- MDCG 2020-6 Regulation (EU) 2017/745: Clinical evidence needed for medical devices previously CE marked under Directives 93/42/EEC or 90/385/EEC.
In tale contesto, appare chiaro che i requisiti normativi da rispettare risultano molteplici e necessitano di tempo, e risorse adeguate a definire e attuare una strategia appropriata. Rivolgersi ad esperti nella progettazione e conduzione di studi può essere di notevole aiuto per interpretare correttamente le disposizioni contenute nel MDR.

























