




Le aziende che hanno dotato la propria organizzazione di un Sistema di Gestione per la Qualità (SGQ) conforme alla norma tecnica ISO 13485 conoscono molto bene il concetto di “validazione”. La norma, al punto 7.5.6, infatti, specifica che “l’organizzazione deve validare tutti i processi per la produzione e l’erogazione di servizi dove gli elementi in uscita risultanti non possono essere o non sono verificati mediante successivo monitoraggio o misurazione e, di conseguenza, le carenze divengono apparenti solo dopo che il prodotto in uso o il servizio sono stati erogati. La validazione deve dimostrare la capacità di questi processi di conseguire coerentemente i risultati pianificati (…)”. Lo stesso requisito impone alle organizzazioni anche di validare le applicazioni software utilizzate nei processi di produzione e di erogazione dei servizi, mentre il successivo (7.5.7) stabilisce requisiti particolari per la validazione dei processi di sterilizzazione e dei sistemi di barriera sterile.
Vale la pena ricordare che la norma ISO 13485 parla di validazione anche in riferimento alle applicazioni di software di computer utilizzate nel sistema di gestione per la qualità. “Convalida (o validazione) di processo” significa stabilire con prove oggettive che un processo produce costantemente un risultato o un prodotto che soddisfa i requisiti predeterminati. Le organizzazioni che si apprestano a validare i propri processi, quindi, lo fanno per assicurarsi che, all’interno di specificati limiti, questi producano in modo costante prodotti conformi ai requisiti predeterminati, definiti con la progettazione.
La validazione di un processo segue normalmente questi passaggi:
- Sviluppo di un piano di validazione
- Definizione dei requisiti principali della validazione e descrizione del processo
- Definizione dei parametri di processo e dell’output desiderato
- Selezione del metodo e degli strumenti per la validazione
- Creazione di un protocollo valido che contenga i requisiti e i risultati attesi, oltre alle modalità per verificarli
- Esecuzione delle tre fasi della convalida (IQ – OQ – PQ) e documentazione dei risultati in un rapporto di validazione
- Interpretazione dei risultati, che prevede una decisione finale su eventuali ulteriori azioni da intraprendere
- Monitoraggio continuo del processo per determinare eventuali necessità di rivalidazione
Le attività di validazione sono tipicamente effettuate secondo un modello costituito dalle tre fasi “Qualifica di Installazione” (IQ), “Qualifica Operativa” (OQ), “Qualifica delle prestazioni” (PQ). La sequenza cronologica di esecuzione di queste fasi è fondamentale: esse devono sempre essere conseguenti l’una all’altra e mai concomitanti. Vediamo di seguito in cosa consistono.
Qualifica di Installazione
La Qualifica di Installazione (IQ) serve a stabilire che tutti gli aspetti fondamentali di installazione delle attrezzature coinvolte nel processo oggetto di validazione aderiscano ai criteri del progetto approvato e che le raccomandazioni dei fornitori siano state adeguatamente considerate. Questa fase può mancare nel caso in cui le attrezzature siano di utilizzo consolidato o siano state validate in modo separato contestualmente alla loro installazione.
Qualifica Operativa
La Qualifica Operativa (OQ) serve a stabilire i limiti di controllo del processo, ovvero i parametri operativi, e le relative fasi di processo che portano a un prodotto che soddisfa tutti i requisiti predeterminati. In questa fase, i parametri di processo devono essere definiti e testati per assicurare che l’output sia un prodotto che soddisfa i requisiti in tutte le condizioni prestabilite del processo. Nella fase di OQ, in base alle specifiche desiderate del prodotto finale, si stabilisce quali sono i parametri che influenzano tali specifiche: sono, quindi, stabiliti i range entro cui ciascun singolo parametro può variare e quali sono le prove da effettuare per mantenerlo sotto controllo.
Per i processi consolidati, i parametri usati in questa fase sono quelli che derivano dalla routine di processo e che potrebbero eventualmente essere modificati in base agli esiti dei test eseguiti in questa fase.
Per ogni test previsto da questa fase della validazione è necessario stabilire:
- Cosa testare
- Come testarlo e con cosa
- Quanto verificare
- Quando verificarlo
- Chi deve verificare
- I criteri di accettabilità
- I requisiti della documentazione
Alla fine della fase di OQ vengono revisionate o confermate le procedure e le istruzioni operative previste per il processo, le procedure di controllo, pulizia e manutenzione, formazione del personale.
Qualifica delle Prestazioni
La Qualifica delle Prestazioni (PQ) serve a stabilire che il processo, in tutte le condizioni previste, produca costantemente un prodotto che soddisfa tutti i requisiti predeterminati. La PQ dimostra che il processo produce in modo consistente prodotti conformi alle specifiche nelle normali condizioni operative. La fase di PQ è condotta sulla base dei parametri definiti in OQ. I test sono eseguiti su un campione statisticamente significativo di prodotti per assicurare la capacità e la stabilità del processo a lungo termine.
Il protocollo di validazione
Dal punto di vista documentale, le modalità di conduzione della validazione di un processo, comprensive di parametri di test, caratteristiche dell’output del processo, elenco delle attrezzature coinvolte, criteri di accettabilità, ecc., sono raccolte in un protocollo di validazione. Il protocollo deve essere emesso e approvato prima dell’inizio delle attività di validazione. Il protocollo di validazione prende in considerazione e definisce:
- impostazioni del processo (settaggio di ogni parametro di processo; controllo delle interazioni tra i parametri)
- condizioni di configurazione (ad es. presenza di un campione di riferimento, se applicabile, personale richiesto, documentazione richiesta al punto di utilizzo, …)
- requisiti dei macchinari coinvolti
- parametri software
- specifiche delle materie prime ed eventualmente requisiti dei relativi fornitori
- procedura di funzionamento del processo ed istruzioni operative applicabili
- controllo delle modifiche al processo (quali cambiamenti sono ammessi senza che sia necessaria una rivalidazione; modalità di gestione delle deviazioni)
- requisiti di formazione del personale
Il protocollo di validazione non può mai essere definito in base ai risultati: qualora durante la sua esecuzione dovessero verificarsi dei risultati inattesi, questi devono essere gestiti come deviazioni e, a seguito di opportune considerazioni emerse da parte del team di validazione, il protocollo può essere revisionato e rieseguito.
Il report di validazione
I risultati ottenuti mediante le attività effettuate secondo il protocollo, la loro interpretazione e le conclusioni a cui il team di validazione è giunto sono raccolti nel report di validazione. Il report è sempre supportato dai “raw data”, ovvero i dati grezzi raccolti durante le attività di validazione. Il report, inoltre, contiene tutte le eventuali deviazioni risultate dall’esecuzione del protocollo di validazione, ovvero tutti risultati ottenuti che si sono rivelati diversi da quelli attesi, e i riferimenti relativi ai rapporti di gestione di tali deviazioni.
Quali processi validare
La norma ISO 13485 specifica che devono essere validati tutti i processi che non possono essere o non sono verificati mediante successive attività di monitoraggio o misurazione. Questo concetto riconduce ad una linea guida emessa dalla Global Harmonization Task Force dedicata proprio alla validazione dei processi, nella quale viene definito un flusso decisionale utile ad orientare la scelta di quali processi sottoporre a validazione.
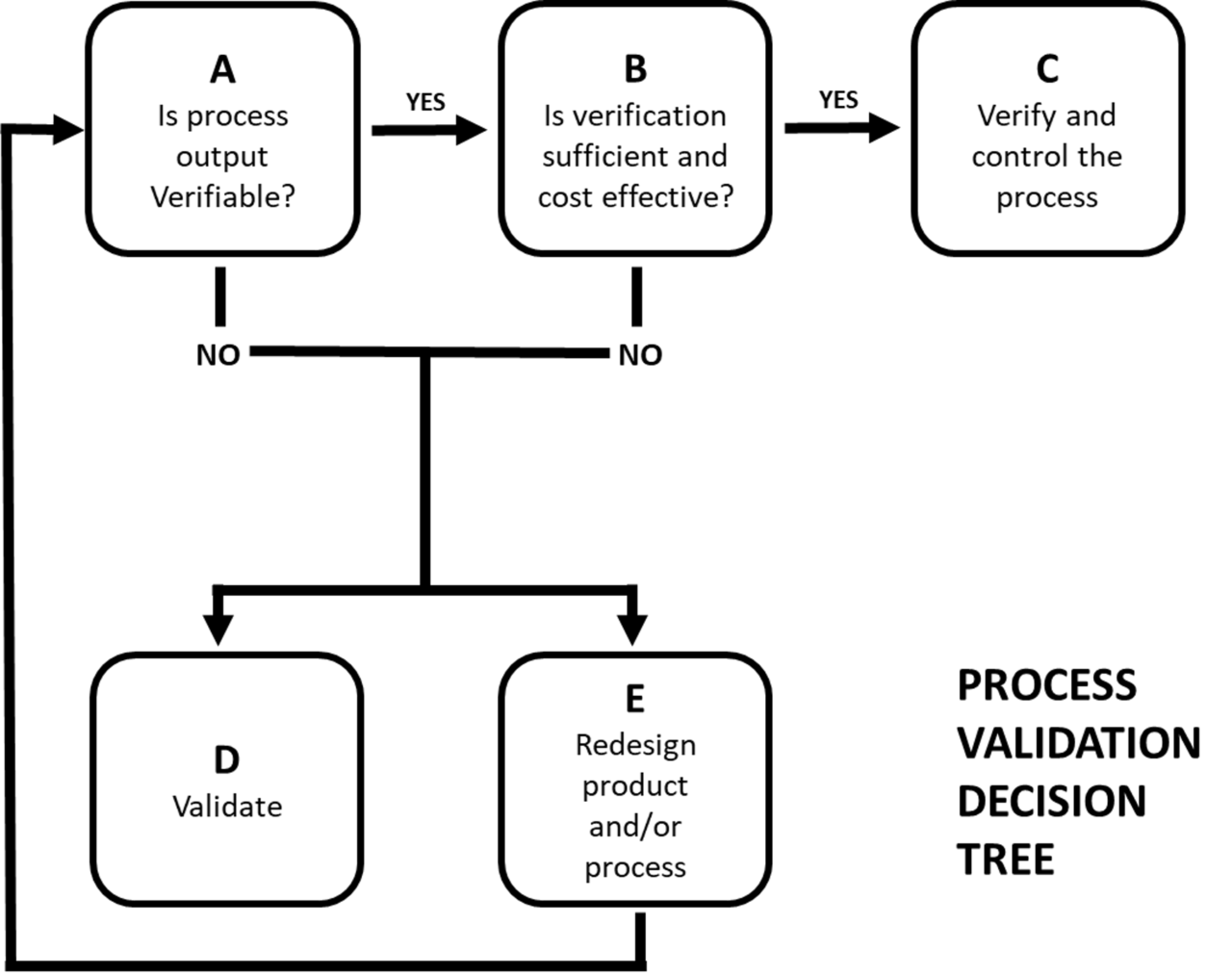
Si presuppone che per ogni processo esista una specifica che descrive sia i parametri del processo sia l’output desiderato di quel processo. Il fabbricante deve considerare se l’output può essere verificato mediante un monitoraggio o una misurazione successivi (A). Se la risposta è positiva, occorre valutare se la sola verifica sia sufficiente ad eliminare i rischi inaccettabili e rappresenti una soluzione economicamente sostenibile (B). In caso affermativo, l’output deve essere verificato e il processo deve essere adeguatamente controllato (C) ma non servirà procedere ad una validazione.
Se, invece, l’output del processo non è verificabile o se la verifica non è sostenibile o non è sufficiente ad abbattere i rischi sino ad un livello accettabile, la decisione dovrebbe essere quella di convalidare il processo (D); in alternativa, potrebbe risultare che il prodotto o il processo debbano essere riprogettati per ridurre i rischi e migliorare il prodotto o il processo (E).
Il team di validazione
Solitamente la responsabilità di gestire le validazioni di processo ricade sul dipartimento qualità dell’azienda, in quanto garante della corretta applicazione dei requisiti normativi. Per condurre l’attività di validazione in modo efficace, tuttavia, è raccomandato che il dipartimento qualità sia costantemente affiancato dai responsabili del processo oggetto di validazione, poiché questi sono i migliori conoscitori del flusso e dei parametri operativi. Si viene quindi normalmente a formare un team di validazione che, con la complementarità delle competenze, riesce a portare a termine tutte le fasi di convalida, documentandole opportunamente.

























