




I dispositivi medici svolgono un ruolo fondamentale nella salute e nel benessere delle persone. Prima che possano essere immessi in commercio negli Stati Uniti devono passare attraverso un rigoroso processo di approvazione. Questo processo, principalmente gestito dalla Food and Drug Administration (FDA), è notoriamente complesso e richiede una pianificazione accurata. I principali riferimenti normativi sono il Federal Food, Drug, and Cosmetic Act (FD&C Act) e le Parti 1-58, 800-1299 del Titolo 21-Codice dei regolamenti federali (21 CFR).
In questo articolo esaminiamo i diversi tipi di richieste di approvazione (submission) per l’immissione in commercio dei dispositivi medici negli Stati Uniti, al fine aiutare gli operatori del settore a orientarsi in questo processo.
L’importanza dell’approvazione della FDA
Perché l’approvazione della FDA è cruciale per immettere in commercio i dispositivi medici? La FDA è l’autorità di regolamentazione negli Stati Uniti responsabile di garantire che i dispositivi medici siano sicuri ed efficaci per l’uso umano. Senza l’approvazione della FDA un dispositivo medico non può essere commercializzato o venduto legalmente negli Stati Uniti. Il processo di approvazione della FDA è rigoroso e consiste in una valutazione completa della sicurezza e dell’efficacia del dispositivo.
I quattro passaggi chiave
Elenchiamo i quattro passaggi chiave per impostare la submission e seguirne l’iter.
- Fase uno: classificare il dispositivo e comprendere i controlli normativi applicabili
- Fase due: selezionare e preparare la corretta Premarket Submission
- Fase tre: inviare la richiesta di pre-immissione in commercio alla FDA e interagire con il personale della FDA durante la revisione della documentazione
- Fase quattro: rispettare i controlli normativi applicabili (inclusa la registrazione dello stabilimento e dell’elenco dei dispositivi)
Fase uno: classifica il tuo dispositivo e individua i controlli applicabili.
Il primo passo è determinare come la FDA ha classificato il tuo dispositivo.
La definizione di dispositivo medico secondo il quadro normativo statunitense è data nella sezione 201(h) della legge federale su alimenti, farmaci e cosmetici (FD&C). Esistono 3 classi di rischio: I, II e III. All’aumentare della classe di rischio aumentano i controlli normativi e i tipi di submission previsti, come riepilogato nella tabella che segue.
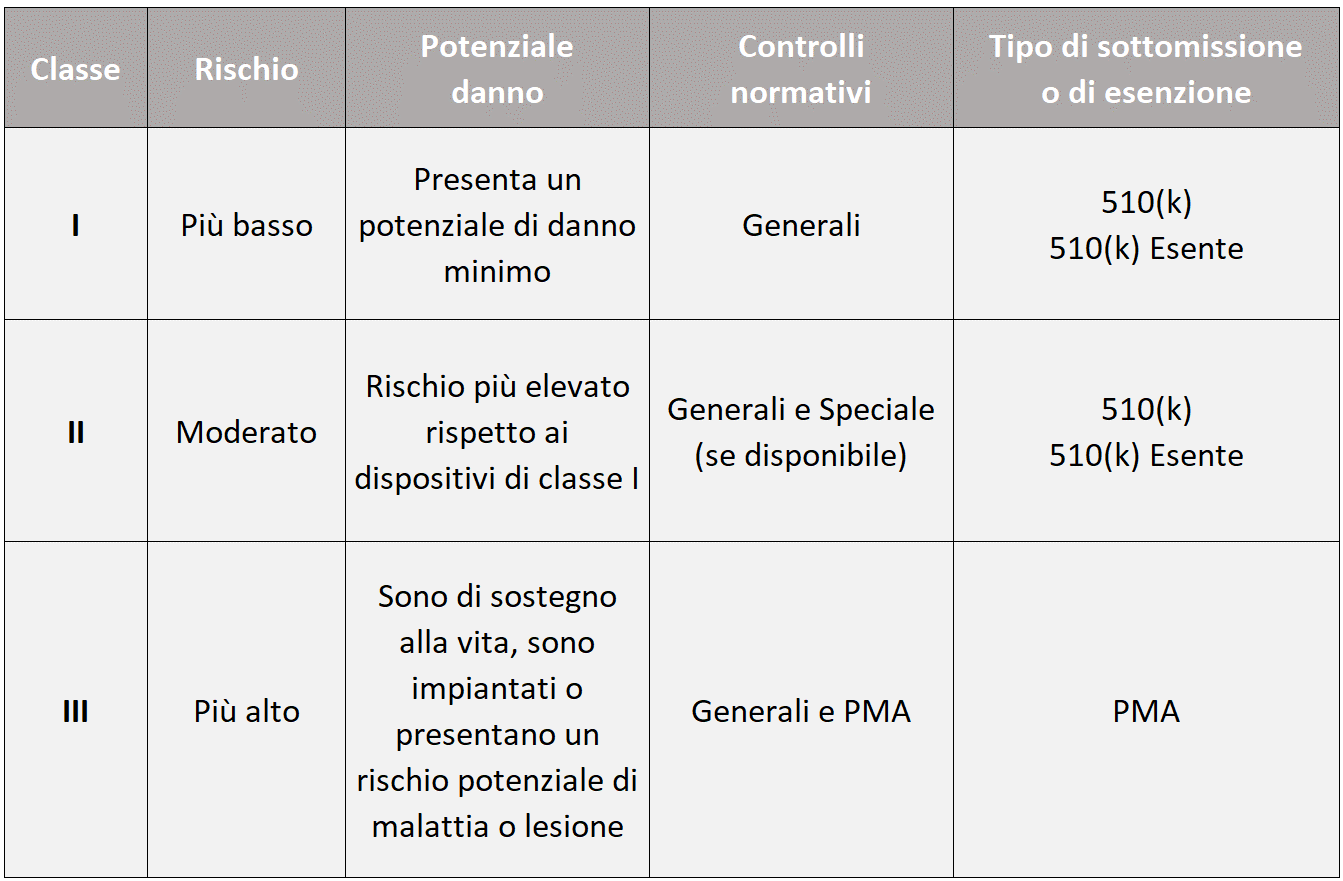
Fase due: selezionare e preparare la corretta Premarket Submission.
Fermo restando che alcuni tipi di dispositivi non richiedono una Premarket Submission (informazioni utili in questo senso possono essere reperite in questa sezione del sito web della FDA Class I/II Exemptions), nella maggior parte dei casi questa è necessaria e il tipo appropriato viene identificato in relazione alla classificazione del prodotto, che può essere ottenuta dal Product Classification database.
I tipi di submission sono sostanzialmente quattro.
- 510(k) (Premarket Notification)
- PMA (Premarket Approval)
- De Novo Classification Request
- HDE (Humanitarian Device Exemption)
I due tipi di submission più utilizzati sono la 510(k) Premarket Notification e la Pre-market Approval (PMA).
La richiesta di Classificazione De Novo è un percorso che viene attivato solo nel caso (si veda anche il link DeNovo Classification Request (De Novo Process) in cui si verificano contemporaneamente le seguenti condizioni:
- è possibile dimostrare l’efficacia e la sicurezza del prodotto solo attraverso i controlli generali o generali e speciali; e
- non esiste un dispositivo simile già approvato (predicate).
La richiesta HDE è un caso particolare in cui l’immissione in commercio è richiesta per dispositivi di classe III destinati a favorire i pazienti affetti da malattie o condizioni rare.
Vediamo nel dettaglio le due opzioni principali, 510(k) (Premarket Notification) e PMA (Premarket Approval)
1. 510(k) Premarket Notification
La submission 510(k) è una delle vie più comuni per ottenere l’approvazione. È impiegata per dispositivi che sono simili a dispositivi già approvati dalla FDA e che sono considerati “equivalenti” da un punto di vista tecnico e di sicurezza. La submission 510(k) richiede all’azienda di dimostrare che il nuovo dispositivo è sostanzialmente equivalente (SE) al dispositivo di riferimento precedentemente approvato attraverso la presentazione di dati clinici, tecnici e scientifici. Questo processo può richiedere tempo e risorse significativi, ma è spesso più rapido ed economico rispetto alla PMA.
2. Pre-Market Approval (PMA)
La submission PMA è richiesta per i dispositivi medici nuovi, innovativi o considerati ad alto rischio. Questa è la strada da percorrere se un dispositivo non può essere classificato come “equivalente” ai dispositivi già approvati. La submission PMA richiede una valutazione completa dei dati clinici, preclinici e scientifici, nonché una dimostrazione rigorosa dell’efficacia e della sicurezza del dispositivo.
Poiché la submission PMA prevede una valutazione più approfondita rispetto alla 510(k), è generalmente un processo più lungo e costoso. Tuttavia, è l’unica via percorribile per valutare i dispositivi che rappresentano una vera e propria innovazione nel campo medico.
Fase tre: inviare la richiesta di pre-immissione in commercio alla FDA e interagire con il personale della FDA durante la revisione – Processo di Submission.
La submission consiste nell’invio della richiesta di pre-immissione in commercio alla FDA e l’interazione con il personale della FDA durante la revisione.
Indipendentemente dal tipo di submission, il processo di approvazione della FDA richiede l’esecuzione di ulteriori passaggi; descriviamo qui di seguito quelli principali.
1. Raccolta di Dati e Progettazione dello Studio
Sia per la 510(k) che per la PMA è necessario raccogliere dati scientifici e clinici per dimostrare l’efficacia e la sicurezza del dispositivo. Questo può includere l’esecuzione di studi clinici, test di laboratorio e prove precliniche. La progettazione di qualunque tipo di studio è cruciale per assicurare che i dati raccolti siano validi e conformi alle linee guida della FDA.
2. Preparazione della Submission
La preparazione della submission richiede la creazione di un dossier che include tutti i dati, i risultati degli studi, le informazioni sulla sicurezza e l’efficacia del dispositivo, e altro ancora. Questo dossier deve essere accurato e ben organizzato, in quanto verrà esaminato nel dettaglio dalla FDA.
3. Invio della Submission
Una volta completata la submission, questa deve essere inviata alla FDA per la revisione. Il processo di revisione può richiedere diversi mesi, durante i quali la FDA esaminerà attentamente tutti i dati presentati.
4. Risposta e Negoziazione
La FDA può fornire una risposta iniziale alla submission, che potrebbe includere richieste di ulteriori informazioni o chiarimenti. Questo processo può articolarsi in molteplici scambi di informazioni tra l’azienda e la FDA per garantire che tutte le preoccupazioni siano affrontate in modo soddisfacente.
5. Approvazione
Una volta che la FDA è soddisfatta dei dati presentati e delle prove fornite, il dispositivo medico può essere approvato per la commercializzazione negli Stati Uniti. L’azienda può quindi iniziare la produzione e la distribuzione del dispositivo.
Fase quattro: rispettare i controlli normativi applicabili (inclusa la registrazione dello stabilimento e l’elenco dei dispositivi).
I controlli normativi sono dei requisiti basati sul rischio che si applicano ai dispositivi medici e che permettono alla FDA di supervisionare e garantire la ragionevole sicurezza ed efficacia dei dispositivi medici.
I dispositivi di tutte e tre le classi sono soggetti a controlli generali (salvo esenzioni) che richiedono che i fabbricanti di dispositivi medici:
- registrino i propri stabilimenti ed elenchino i dispositivi medici che commercializzano presso la FDA;
- fabbrichino i propri dispositivi in conformità con le buone pratiche di fabbricazione;
- etichettino i propri dispositivi in conformità con le normative applicabili; e
- non adulterino o marchino in modo errato i dispositivi.
Se un dispositivo richiede una pre-market submission prima della commercializzazione (vale a dire, il dispositivo medico non è esente), il fabbricante deve attendere l’autorizzazione o l’approvazione della FDA prima della registrazione e del “device listing”.
Considerazioni Chiave
Durante il processo di submission per l’approvazione della FDA, ci sono diverse considerazioni chiave da tenere presente.
- Il processo di submission richiede una pianificazione accurata e una gestione del tempo efficace. È importante iniziare la pianificazione molto prima della submission effettiva, in modo da poter raccogliere i dati necessari e condurre gli studi clinici o preclinici, se necessario.
- È essenziale che tutte le fasi della submission siano conformi alle normative della FDA. L’azienda deve essere a conoscenza di tutte le leggi e i regolamenti applicabili e assicurarsi di rispettarli rigorosamente.
- Il processo di submission può essere costoso. Le aziende devono essere pronte a investire risorse finanziarie significative nella raccolta dei dati e nella gestione del processo di approvazione.
- La submission richiede un’ampia conoscenza delle normative e dei requisiti della FDA. Molte aziende scelgono di collaborare con esperti regolatori o consulenti per garantire che il processo sia gestito correttamente.
L’approvazione della FDA è un passaggio cruciale per poter immettere in commercio i propri dispositivi medici nel mercato statunitense. Identificare il tipo di submission adeguata, redigere accuratamente il dossier di presentazione, interagire efficacemente con FDA sono tutti passaggi fondamentali: per questo, se non si è sicuri di essere in grado di procedere in autonomia, affidarsi a esperti regolatori e legali può essere la scelta opportuna per garantire una navigazione senza intoppi attraverso il processo di approvazione della FDA. Una pianificazione accurata e una gestione competente possono aumentare notevolmente le probabilità di successo per l’immissione sul mercato statunitense dei dispositivi medici.























